(Terapia celular en neuropatología)
(Terapia celular en neuropatología)
Duración: 2 años.
Coste del proyecto: 100.000€ por cada modelo animal/sindrome
Manuel Álvarez Dolado

OBJETIVO
El objetivo principal del presente proyecto es desarrollar una nueva terapia celular mediante el trasplante de progenitores de interneuronas GABAérgicas derivados de la MGE en modelos murinos de Encefalopatias Epilépticas Infantiles como los Síndrome de Dravet, de West y el Stxbp1. El proyecto recopilará la información preclínica necesaria para analizar los beneficios de esta terapia celular y confirmará su posible aplicación clínica en un futuro. Para ello se analizarán los efectos del trasplante a nivel electrofisiológico, histológico, molecular y de comportamiento.
ANTECEDENTES Y ESTADO ACTUAL DEL TEMA
La epilepsia es una afección crónica, de etiología muy diversa, debida a un desequilibrio entre excitación e inhibición que conduce a una hiperactividad neuronal incontrolada. En general, las crisis pueden deberse a una hiperactividad de las neuronas excitatorias o, por el contrario, a la ausencia de actividad de las interneuronas que conforman el sistema inhibitorio, normalmente por alteraciones en las neuronas GABAérgicas. Son numerosas las evidencias que relacionan estrechamente GABA y epilepsia. De ahí que muchas drogas tengan como diana el sistema GABAérgico. Sin embargo, pese a la eficacia comprobada de estos fármacos, entre los pacientes epilépticos existe un grave problema de farmacorresistencia. Se calcula que alrededor del 30% de las epilepsias son incontrolables con tratamiento farmacológico. Esta cifra puede llegar hasta un 70% de los pacientes con determinados tipos de epilepsia clasificadas como raras por su baja frecuencia. Entre las epilepsias refractarias raras destacan síndromes de origen genético de inicio temprano, como las encefalopatías epilépticas infantiles tempranas (EIEE). Estas enfermedades están pobremente caracterizadas y carecen de tratamiento farmacológico efectivo, bien definido y sin efectos secundarios graves, por lo que es necesario el desarrollo de nuevas alternativas terapéuticas.
Las EIEE se caracterizan por tres criterios diagnósticos principales: crisis médicamente refractarias, encefalopatía difusa y fuerte retraso en el desarrollo psicomotor. En esta clasificación se incluyen el Síndrome Stxbp1; el de espasmos epilépticos infantiles ligado al cromosoma-X, también conocido como Síndrome de West (SW); y el Síndrome de Dravet (SD). Estos últimos se caracteriza por una elevada mortalidad asociada a la epilepsia con una edad de fallecimiento muy temprana, el 10-18% de los pacientes muere entre los 3 y 7 años de edad por muerte súbita e inesperada en la epilepsia (SUDEP).
Las causas de estos síndromes están en mutaciones de determinados genes que guardan relación con el correcto funcionamiento del sistema GABAérgico. Así, el SD se debe a pérdidas de función por mutaciones en heterocigosis del gen SCN1a, que es fundamental para la correcta despolarización de las interneuronas inhibitorias GABAérgicas. Mientras que el SW es causado por mutaciones en el factor de transcripción ARX, que es esencial para una adecuada diferenciación y desarrollo de las interneuronas corticales y del hipocampo. Por último, el Síndrome Stxbp1 se debe a mutaciones en el gen que codifica para Munc18-1, una proteína fundamental para la correcta transmisión sináptica.
Nuestro laboratorio dispone de ratones transgénicos nulos y condicionales para estos genes, lo que nos permite ensayar drogas y nuevas aproximaciones terapéuticas para tratar estas enfermedades. Una posible alternativa es la terapia celular de reemplazo, que podría sustituir las interneuronas que no funcionan adecuadamente. Nuestro grupo viene trabajando en este tipo de terapia en los últimos años, en el marco de varios proyectos financiado anteriormente por diferentes entidades, como el Plan Nacional, la Junta de Andalucía o la Fundación Ramón Areces.
La terapia celular ha surgido en los últimos años como una posibilidad para no solo paliar, sino curar, diversas enfermedades que afectan al sistema nervioso, entre ellas la epilepsia. Varios grupos, incluyendo el nuestro, venimos trabajado desde hace años en una nueva fuente de precursores GABAérgicos con resultados muy alentadores. Se trata de progenitores neuronales que se originan en la eminencia ganglionar medial (MGE).
Esta región da lugar a las interneuronas GABAérgicas del cortex y el hipocampo durante el desarrollo normal del cerebro. Nuestro grupo es capaz de aislar estos progenitores y realizar trasplantes en el cerebro adulto y neonato (P3-5). Hemos observado que las células de la MGE al ser trasplantadas migran varios milímetros de distancia alrededor del lugar de inyección, llegando a cubrir la práctica totalidad del hipocampo y cortex cerebral con una sola inyección (Fig. 1). Su supervivencia es alta y en 3-4 semanas se diferencian de forma natural hacia interneuronas GABAérgicas maduras plenamente funcionales, demostrado mediante la expresión de marcadores y análisis electrofisiológico de sus propiedades de disparo. Su diferenciación conlleva la modulación de la actividad inhibitoria cerebral en la zona del trasplante, recuperando el tono inhibitorio necesario para frenar las crisis epilépticas.
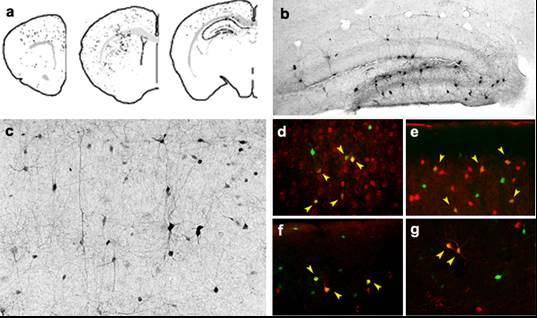
Numerosas publicaciones sobre trasplantes en diferentes modelos animales de neuropatologías relacionadas con las interneuronas confirman el gran potencial terapéutico que poseen este tipo de progenitor neuronal. Destacar especialmente nuestro trabajo recientemente publicado en Neuron en el que demostramos que el trasplante en un modelo de Alzheimer caracterizado por el déficit en la expresión de Nav1.1, similar a lo que ocurre en el SD, recupera las alteraciones cognitivas y los ritmos cerebrales que sufre este modelo. El análisis del espectro de ondas de los EEG indica una normalización de la ritmogénesis cerebral en estos animales gracias al trasplante, lo que conlleva una reducción en sus comportamientos hiperactivos, ansiedad y recuperación de la cognición. Por otra parte, ensayos ya avanzados en el modelo de SW demuestran recuperación del fenotipo ansioso e hiperactivo de los ratones en las pruebas de comportamiento y una reducción significativa en la frecuencia de los espasmos epilépticos. Todos estos datos sugieren fuertemente la hipótesis de que una terapia celular con progenitores GABAérgicos derivados de la MGE puede ser beneficiosa para el SD o en otras EIEE como el SW y el de Stxbp1.
En su conjunto, el proyecto profundizará en nuestro conocimiento sobre las encefalopatías epilépticas infantiles de inicio temprano y completar los ensayos pre-clínicos en modelos animales de una novedosa estrategia terapéutica basada en células. Con ello afrontamos el reto de comprender y desarrollar nuevos tratamiento para unas patologías tan devastadoras, que afectan a niños y que no tienen solución a día de hoy.
Metodología y Fases
Para alcanzar este objetivo el proyecto se dividirá en 4 fases, de 6 meses de duración cada una, en las que se establecerán una serie de hitos a alcanzar para continuar con el proyecto. Al finalizar cada fase se realizarán reuniones y memorias de seguimiento en donde se informará del nivel de cumplimiento de estos hitos, los resultados obtenidos y posibles dificultades que nos encontremos. A continuación se describen los hitos a conseguir en cada fase:
Fase 1
- Obtener una colonia suficientemente grande de ratones modelo para realizar todos los ensayos futuros.
- Tener una caracterización a nivel de comportamiento y de EEG de los ratones modelo.
- Preparar los cruces para las cohortes de animales modelo y de donantes con los que realizar el trasplante neonatal durante la siguiente fase.
Fase 2
- Realizar los trasplantes en ratones neonatos.
- Obtener información de los beneficios del trasplante a nivel de comportamiento.
- Disponer de todo lo necesario para realizar el trasplante en adulto durante la siguiente fase
Fase 3
- Comprobar los beneficios del trasplante neonatal a nivel electrofisiológico (EEG, reducción de crisis, espectro de ondas).
- Iniciar la toma de muestras de la cohorte neonatal.
- Realizar los trasplantes en ratones adultos.
- Realizar los test de comportamiento en la cohorte adulta.
Fase 4
- Comprobar los beneficios del trasplante adulto a nivel electrofisiológico y de conducta.
- Toma de muestras de la cohorte adulta y análisis histológico y molecular junto a la neonatal.
- Análisis global del proyecto, conclusiones e informe final.
Contratación personal con dedicación exclusiva
Mantenimiento colonia de animales modelo
Instituciones colaboradoras
CABIMER
Entidades financiadoras
Ministerio de Educación y Ciencia, Junta de Andalucía, Fundación Ramón Areces, Apoyo Dravet